Pneumologie
Fibrose pulmonaire idiopathique : la physiopathologie liée à une anomalie de la ciliogénèse
Une anomalie de la ciliogénèse interviendrait dans le mécanisme physiopathologique de la fibrose pulmonaire idiopathique. Des résultats fondamentaux intéressants mais dont l’applicabilité nécessite d’aller plus loin. C’est une première pierre à l’édifice. D’après un entretien avec Mada GHANEM.

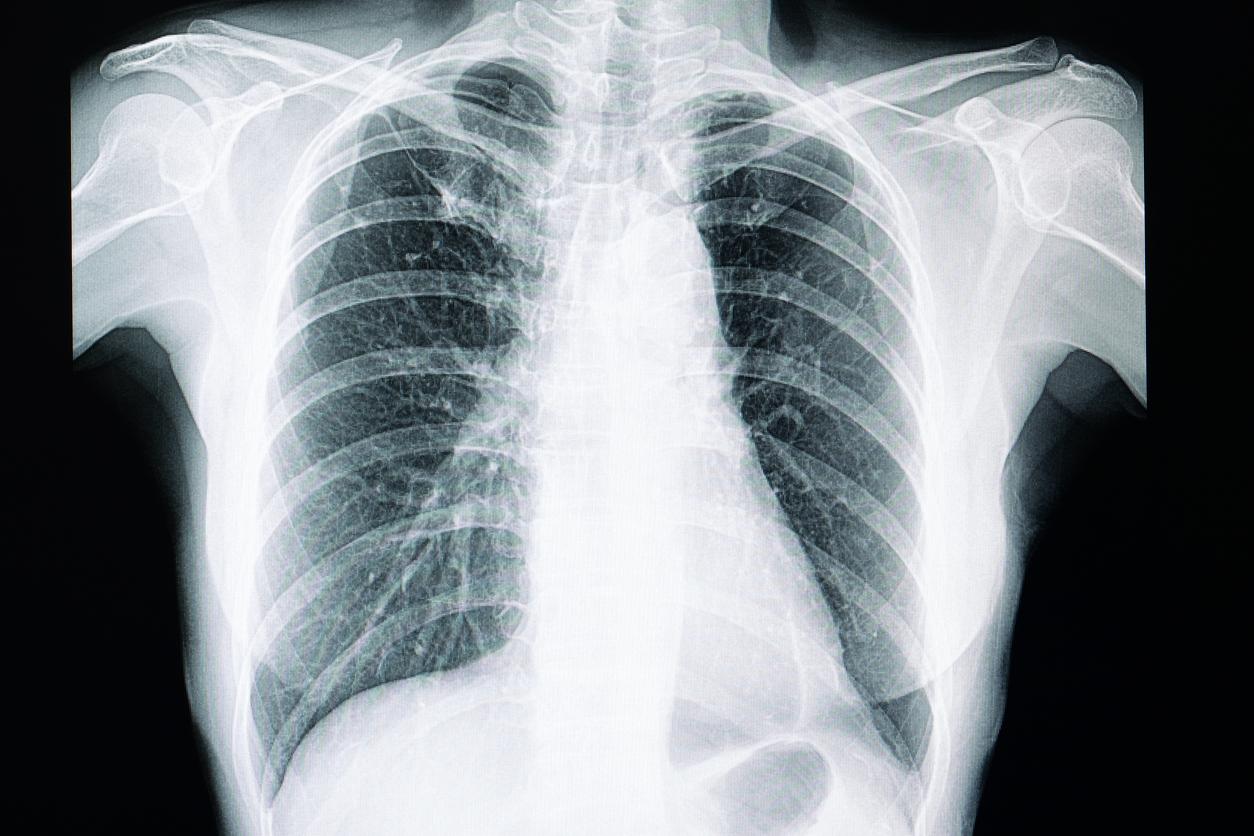
Une étude, dont les résultats sont parus en mai 2022, dans l’American Journal of Respiratory Cell and Molecular Biology, a cherché à montrer l’association du gène MUC5B avec l’expression de la ciliogénèse dans les cellules des voies respiratoires pour expliquer le mécanisme physiopathologique de la fibrose pulmonaire idiopathique. Les auteurs émettent l’hypothèse que cela représenterait une justification supplémentaire du rôle de la ciliogénèse dans la physiopathologie de la fibrose pulmonaire idiopathique. Ils ont donc démontré qu’une augmentation aberrante de la ciliogénèse, par expression accrue du gène MUC5B dans les progéniteurs des cellules Krt5 expliquerait le développement d’une fibrose, hypothèse appuyée par les modèles murins dans lesquels la fibrose pulmonaire est induite par la bléomycine, qui a pour propriété d’augmenter le nombre des cellules Krt5.
Une implication des cellules épithéliales dans le développement de la FPI
Le docteur Mada GHANEM, pneumologue dans le service de Pneumologie et de Maladies pulmonaires rares de l’hôpital Bichat à Paris et auteur de l’éditorial accompagnant cette publication, réalise actuellement une thèse de sciences sur la fibrogénèse pulmonaire. Elle explique que la physiopathologie de la fibrose pulmonaire idiopathique est liée à des lésions répétées de l’épithélium alvéolaires, avec une destruction progressive du parenchyme. Il s’agit d’un mécanisme de bronchiolisation au cours duquel l’épithélium alvéolaire est détruit et remplacé par des cellules épithéliales. Mada GHANEM rappelle qu’il existe plusieurs modèles murins qui ont permis aux auteurs d’émettre l’hypothèse qu’une ciliogénèse aberrante par anomalie génétique pourrait intervenir dans la fibrogénèse. En particulier, le gène MUC5B pourrait être impliqué. Il est associé à des facteurs de transcription de la ciliogénèse et est davantage présent chez les sujets atteints de fibrose que chez les sujets sains, ce qui suggère une atteinte des cils dans la fibrose pulmonaire idiopathique. La présence de MUC5B en quantité élevée est un facteur de risque de développement de FPI connu mais le mécanisme physiopathologique ne l’était pas. Il provoquerait une altération de la fonction mucociliaire. Les cellules Krt5 sont des progéniteurs de la fibrose. Dans les modèles murins, la FPI est induite par la bléomycine qui augmente la population de cellules Krt5. L’expression du gène MUC5B dans les cellules progénitrices de Krt5 serait une cible thérapeutique potentielles pour la FPI.
Des résultats fondamentaux, non encore applicables
Mada GHANEM souligne que la méthodologie de ce travail est intéressante mais que les résultats restent fondamentaux et loin d’être applicables. Ils s’intègrent dans le spectre de la physiopathologie de la fibrose pulmonaire interstitielle, comme une première pierre à l’édifice. Pour Mada GHANEM, il est nécessaire d’aller plus loin pour envisager ce mécanisme comme une éventuelle cible pour un nouveau traitement. Elle précise que plus il y aura de données précises, plus on développera de nouvelles thérapies. De nouveaux travaux sur le sujet doivent donc être réalisés pour comprendre parfaitement la physiopathologie de la fibrose pulmonaire interstitielle et utiliser certains mécanismes comme des leviers thérapeutiques.
En conclusion, les anomalies de la ciliogénèse représentent une partie du mécanisme physiopathologique de la fibrose pulmonaire idiopathique. C’est un premier pas qui ouvre la voie vers de nouvelles connaissances qui pourront permettre d’imaginer des thérapeutiques spécifiques et efficaces.



















, comprendre pour mieux prévenir")






")
