Rhumatologie
Ostéomalacie oncogénique : douleurs, faiblesse musculaire et hypophosphorémie isolée
L'ostéomalacie induite par une tumeur (OIT), également connue sous le nom d'ostéomalacie oncogénique, est une forme acquise d'hypophosphatémie, ou hypophosphorémie, habituellement associée à des tumeurs mésenchymateuses, le plus souvent bénignes. La résection de la tumeur entraîne la guérison de l'ostéomalacie.
- Springsky/istock
L'ostéomalacie induite par une tumeur (OIT) est un syndrome paranéoplasique rare généralement causé par des tumeurs de petite taille, à croissance lente et le plus souvent bénignes, à savoir les tumeurs mésenchymateuses phosphaturiques (TMP).
L'ostéomalacie induite par les tumeurs (OIT) est un trouble acquis de perte rénale isolée de phosphore. Les patients atteints d'OIT ont des phénotypes biochimiques et squelettiques similaires à ceux des patients souffrant de rachitisme hypophosphatémique autosomique dominant (ADHR) et d'hypophosphatémie liée à l'X, ce qui impose une enquête familiale systématique à la recherche d’antécédents familiaux, avant d’en poser le diagnostic.
L'OIT se caractérise cliniquement par des douleurs osseuses du bassin ou plus diffuses, une faiblesse musculaire proximale et des fractures, une perte rénale de phosphore avec hypophosphorémie, alors que les concentrations sériques de 1,25(OH)2D sont faibles ou normales et que les taux sériques de phosphatase alcaline sont élevés. Le facteur de croissance des fibroblastes 23 (FGF-23), une « phosphatonine », a été identifié comme un facteur majeur responsable de la déperdition isolée de phosphore dans l'OIT.
Un syndrome paranéoplasique lié surtout à une hypersécrétion de FGF23
Les tumeurs mésenchymateuses phosphaturiques sont généralement des néoplasmes polymorphes de petite taille, bénins et à croissance lente, affectant les os ou les tissus mous. Elles sécrètent le « facteur de croissance des fibroblastes 23 » (FGF23) et, rarement, d'autres phosphatonines, comme : la « protéine sécrétée liée à la protéine frizzled-4 », le « FGF7 » et la « phosphoglycoprotéine extracellulaire matricielle » (MEPE)
Physiologiquement, la sécrétion de FGF23 est stimulée par le phosphore, l'hormone parathyroïdienne (PTH), les cytokines inflammatoires et le 1,25(OH)2D3. Le FGF23 diminue le seuil de réabsorption tubulaire du phosphore en réduisant l'expression tubulaire du cotransporteur de phosphate de sodium de type II (NPT-2a) et inhibe l'activité biologique de la 1-alpha hydroxylase, empêchant l'activation de la 25-hydroxy-vitamine D (25OHD) en 25-dihydroxy-vitamine D (1,25(OH)2D3).
En conséquence, la sursécrétion de FGF23 par les tumeurs mésenchymateuses phosphaturiques (TMP) induit une fuite rénale isolée de phosphore, avec une hypophosphorémie sévère (< 0,8 mmol/L), une 1,25(OH)2D3 faible ou anormalement normale et, à long terme, une véritable ostéomalacie.
Un syndrome clinique d’ostéomalacie
Les signes et symptômes cliniques liés à l'ostéomalacie induite par une tumeur (OIT) sont une faiblesse musculaire (voire un véritable déficit musculaire) prédominant aux racines, et pouvant aller jusqu’à des troubles de la marche avec démarche dandinante (« en pingouin »). Il existe des douleurs osseuses, prédominant au bassin avec douleurs pelviennes et crurales antérieures, mais aussi des douleurs de la colonne dorsale et des jambes. Ces douleurs sont permanentes ou réveillées par les mouvements et les régions atteintes sont douloureuses au toucher. Des fractures pathologiques et/ou des pseudo-fractures et des déformations de la colonne en cyphose peuvent se produire.
Sur le plan biochimique, il est impossible de distinguer cette maladie des formes héréditaires de rachitisme hypophosphatémique ; il est donc essentiel d'étudier soigneusement les antécédents familiaux ce qui ferait pencher le diagnostic en faveur de ces formes héréditaires.
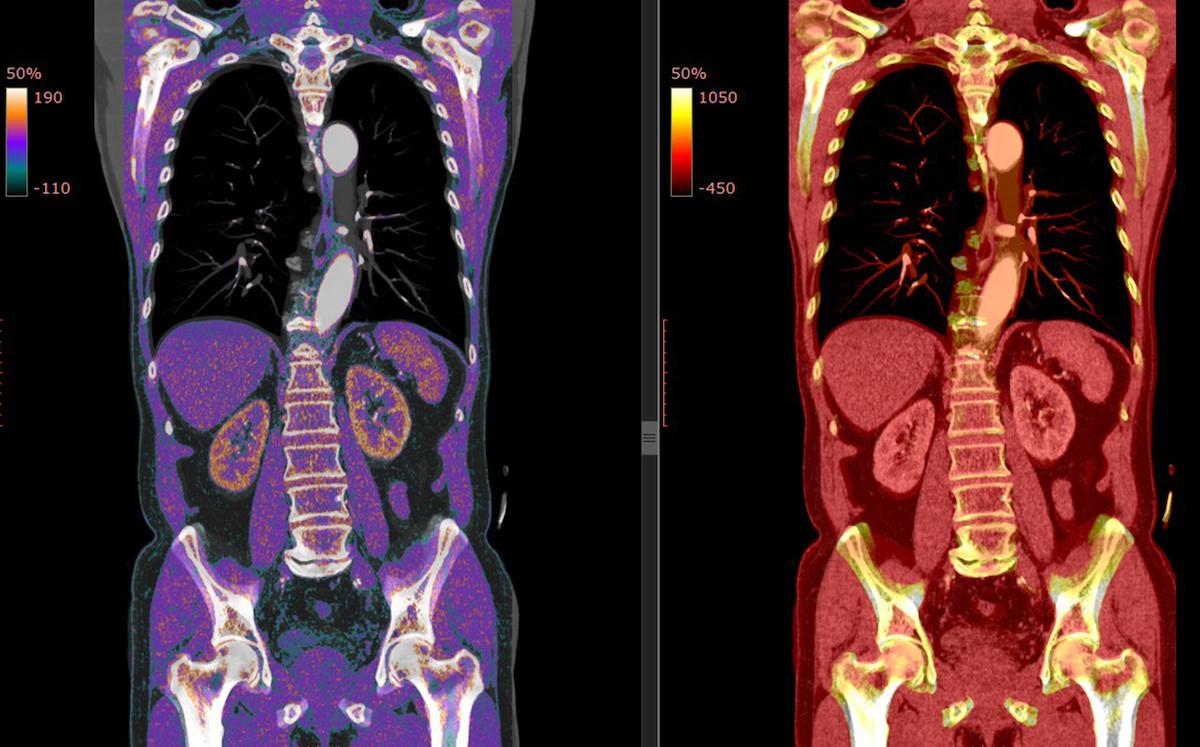
L'ablation chirurgicale des tumeurs mésenchymateuses phosphaturiques permet une rémission complète du syndrome lié à l'OIT dans plus de 90% des cas. Par contre, les TMPs sont généralement difficiles à localiser, et les techniques d'imagerie hybride, telles que la tomographie par émission monophotonique (CT) au 99mTc HYNIC-TOC et la TEP-CT au 68Ga-DOTATOC, se sont avérées avoir une sensibilité maximale de 92% et 91%, respectivement. L’IRM complète l’analyse de la tumeur lorsqu’elle est localisée. Lorsque le néoplasme responsable de l'OIT ne peut être identifié ou éliminé, certaines options ont été proposées, telles que l'utilisation de l'anticorps humain FGF23, le burosumab, et d'analogues de la somatostatine radiomarqués
Une minorité de formes malignes ou évoluant vers une forme maligne
Comparés aux patients souffrant d'ostéomalacie induite par une tumeur (OIT) avec tumeurs mésenchymateuses phosphaturiques (TMP) bénignes, les patients souffrant d’OIT avec TMP maligne auraient des valeurs intactes de FGF23 et de calcium plus élevées et des valeurs plus faibles de phosphatases alcalines, de TmPO4/GFR et de phosphore. Dans une revue de la littérature, les patients souffrant de OIT avec TMP maligne avaient un âge plus bas à l'apparition des signes et symptômes liés à la OIT, un âge plus bas au diagnostic de l’OIT et un taux de mortalité plus élevé que les patients souffrant de OIT avec TMP bénigne.
En outre, certains paramètres (FGF23 intact, calcium sérique, phosphore et TmPO4/GFR) seraient indépendamment associés au diagnostic de TMP maligne, et ils façonneraient le profil de risque de malignité chez les patients atteints d’OIT.
D'après l'analyse multivariée, si les patients atteints d'OIT ont un seul paramètre supérieur (pour le FGF23 intact et le calcium sérique) ou inférieur (pour le phosphate sérique et le TmPO4/GFR) à la valeur médiane calculée pour l'ensemble de la population, le risque d'être atteint d'une PMT maligne est multiplié par plus de 20.
En conclusion
Le diagnostic d'ostéomalacie induite par une tumeur (OIT) peut s'avérer difficile car les tumeurs mésenchymateuses sont souvent petites et difficiles à trouver du fait de leur taille et de leurs localisations parfois atypiques.
La scintigraphie osseuse, la scintigraphie au pentetréotide ou à l'octréotide à l'indium-111 et la tomographie par émission de positons (TEP) sont nécessaires pour tenter de localiser la tumeur et l’analyser par scanner (TDM) ou imagerie par résonance magnétique (IRM) avant son exérèse chirurgicale, qui mettra fin au problème et au risque de dégénérescence.




















")
