Cardiologie
Insuffisance coronaire : des bactéries associées à la rupture des plaques
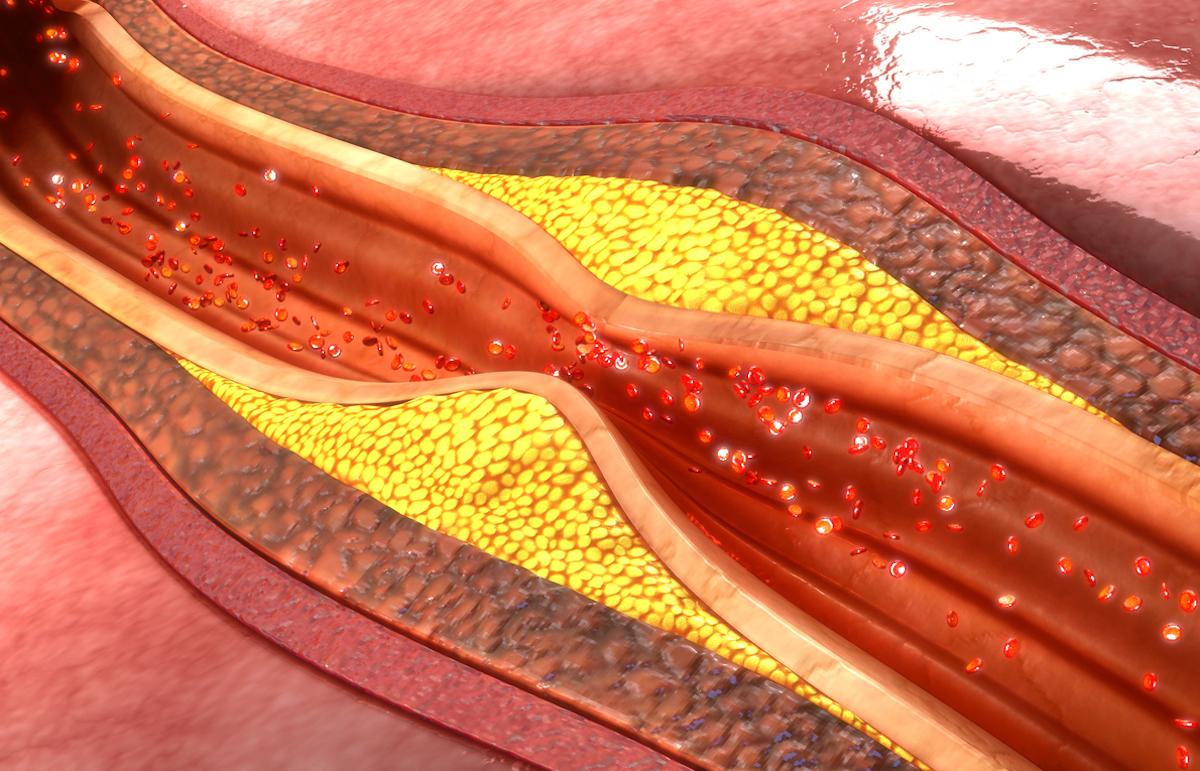
Des bactéries buccales, en particulier les streptocoques du groupe viridans, colonisent les plaques athéromateuses sous forme de biofilm et sont associées à la sévérité des lésions et au risque de mort subite par infarctus. L’activation des voies TLR (surtout TLR2) au contact de bactéries « libérées » du biofilm pourrait contribuer à la rupture de chape fibreuse.
- 7activestudio/istock
L’athérosclérose est une inflammation subclinique associée à l’excès de LDL-cholestérol et impliquant l’immunité innée. Mais l’hypothèse infectieuse refait surface : chez 121 victimes de mort subite et 96 patients opérés d’endartériectomie, de l’ADN bactérien oral était fréquemment détecté au sein des plaques, avec un signal dominant pour les streptocoques viridans.
Selon des données publiées dans le Journal of American Heart Association, en RT-qPCR et immunohistochimie, ces bactéries seraient présentes dans 42,1 % des plaques coronaires et 42,9 % des endartériectomies. Leur immunopositivité serait corrélée à la sévérité de l’athérosclérose (p < 0,0001) et, à l’autopsie, au décès par cardiopathie ischémique (p = 0,021) ou infarctus (p = 0,042). Les Streptococcus viridans coloniseraient le noyau lipidique sous forme de biofilm, discret pour l’immunité innée ; lors d’une rupture, des phénotypes plus virulents infiltreraient la chape fibreuse rompue, seraient reconnus par les TLR et coexisteraient avec une réponse adaptative T (CD3/CD247), scénario compatible avec la transition vers la plaque vulnérable.
De l’inflammation chronique aux signes d’infection latente
En lignées cellulaires, les streptocoques (et certaines bacilles Gram–) activent préférentiellement TLR2 à forte concentration ; dans quelques plaques rompues, TLR4 sont co-exprimé (pneumolysine de S. pneumoniae). In situ, les macrophages CD68+ co-expriment MyD88, NF-κB, TLR2/TLR4 et CD14 au voisinage de la chape fine, soutenant une cascade pro-inflammatoire propice à l’induction de cytokines (IL-6, TNF) et de métalloprotéinases délétères pour le collagène, donc pour l’intégrité de la chape.
L’analyse d’expression globale dans les plaques opérées confirme la surexpression des voies de reconnaissance bactérienne. D’autres signatures microbiennes (Veillonella, Prevotella, Chlamydia pneumoniae 3,8–10,5 %) sont plus rares, suggérant un « microbiome » de plaque parcimonieux ; l’essentiel du signal semble porté par les streptocoques viridans, déjà connus en cas de bactériémie après soins dentaires et dans l’endocardite. Cette étude rappelle également pourquoi les essais d’antibiotiques à large spectre ont échoué :les biofilms sont résistants aux antibiotiques et la dispersion bactérienne est intermittente et peu accessibles à des cures courtes.
Une nouvelle vision de la rupture de plaque
Le travail combine une série médico-légale d’athéromes coronaires (mort subite extrahospitalière) et des spécimens carotidiens/périphériques prélevés en conditions stériles. Trois approches convergentes—RT-qPCR, immunohistochimie bactérienne (anticorps « maison » anti-viridans) et transcriptomique—ancrent l’association entre biofilm streptococcique, activation TLR-dépendante et complications thrombotiques. Des limites imposent la prudence : faible nombre absolu d’événements par espèce hors viridans, impossibilité de distinguer formellement colonisation ancienne versus arrivée récente, hétérogénéité intrinsèque des prélèvements et biais démographiques d’une cohorte d’autopsie. Néanmoins, la concordance entre séries, la colocalisation immunitaire dans les zones de rupture et l’up-régulation des gènes TLR renforcent la plausibilité causale.
Selon les auteurs, ces données confortent une prévention cardio-métabolique qui inclut la santé orale : dépistage et prise en charge des foyers dentaires/periodontaux, information sur le risque de bactériémie transitoire lors des procédures, sans justifier pour autant des antibioprophylaxies hors indications. Elles suggèrent d’être vigilant lors d’infections aiguës (respiratoires, urinaires, cutanées), périodes où la dispersion bactérienne pourrait favoriser l’instabilité de la plaque. Sur le plan thérapeutique, elles ouvrent des pistes : ciblage des biofilms (anti-biofilm, enzymes de matrice, stratégies « quorum sensing »), modulation des voies TLR/MyD88 en complément des traitements hypolipémiants/anti-inflammatoires déjà validés (l’axe IL-1β ayant prouvé sa pertinence dans CANTOS). En somme, l’athérosclérose pourrait bien être, aussi, une maladie de biofilm : l’identifier peut changer la manière de penser la prévention de la rupture de plaque.


















, comprendre pour mieux prévenir")



")
